Most traditional ecological and evolutionary modeling of microbes focuses on steady-state growth. However, it seems unlikely that microbes in nature grow in steady state for very long; changing environments and their sensitive physiology suggest microbes probably undergo frequent shifts in growth. Our work in this research direction has aimed to determine the consequences of non-steady-state growth for ecology and evolution (Fink and Manhart 2023).
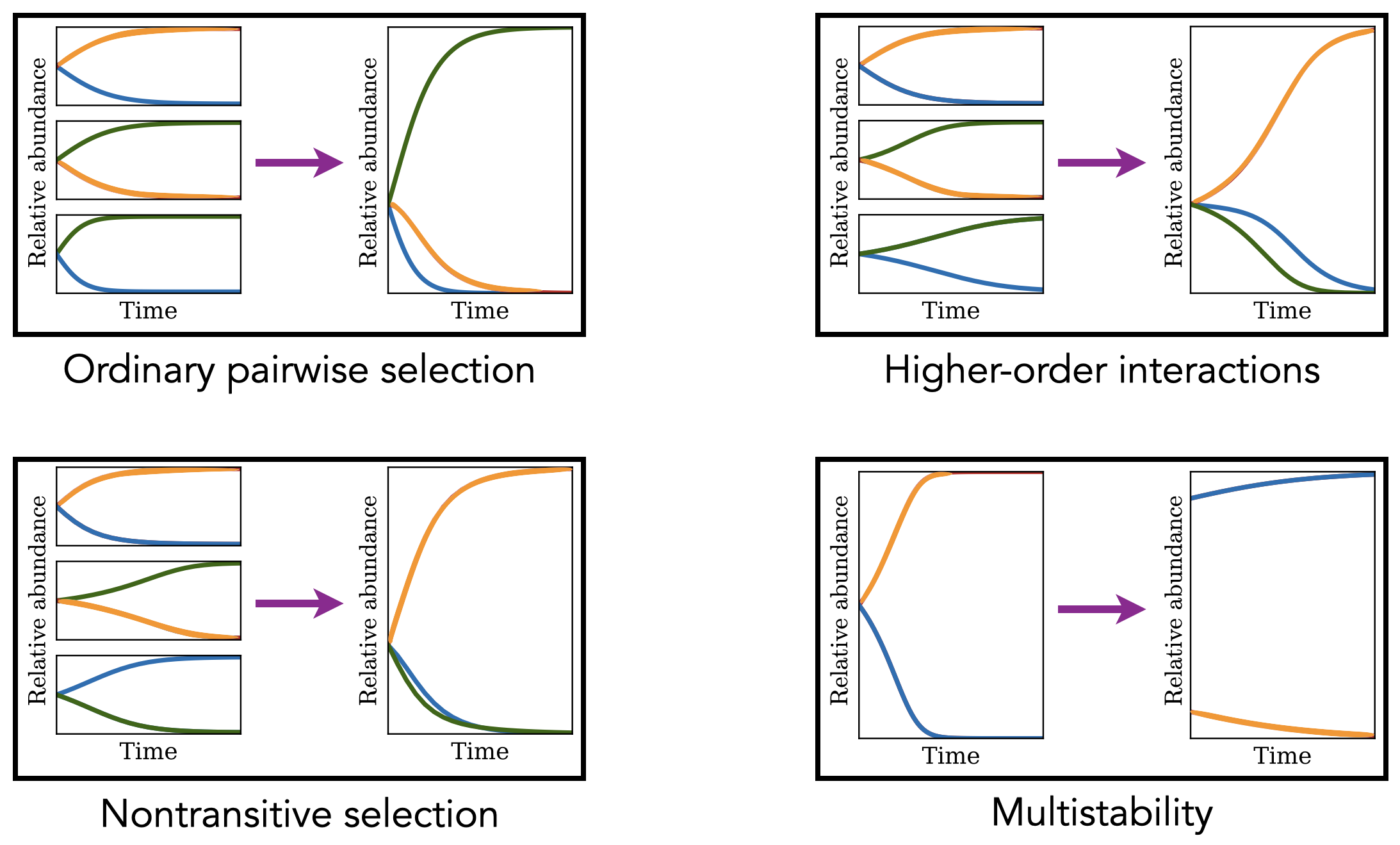
Our earlier work on this topic used mathematical models to show that covariation in microbial growth traits (such as lag times and growth rates) can give rise to complex ecological dynamics, including stable coexistence of multiple species, non-transitive selection, and higher-order ecological effects (Manhart et al. 2018, Manhart and Shakhnovich 2018). Using laboratory growth data, we showed that mutations in microbes are pleiotropic with respect to these traits but do not entail tradeoffs on average (Adkar et al. 2017, Fink et al. 2023, Fink and Manhart 2024), which suggests that complex ecological dynamics are not commonly realized from growth trait variation alone. We have also recently used mathematical models to show how this empirical variation in growth traits affects estimates of fitness from high-throughput experiments (Fink and Manhart 2024).
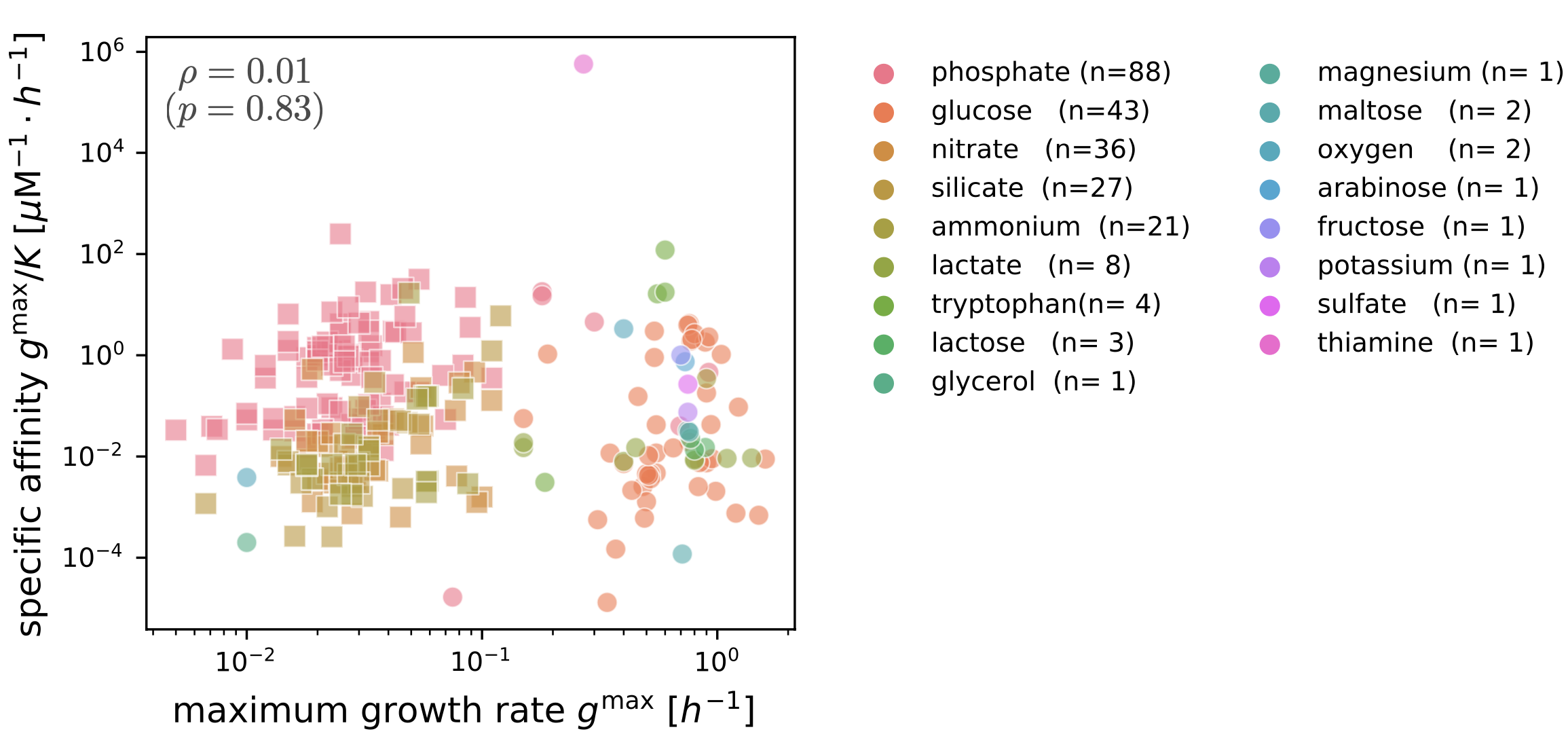
The current focus of this research direction in our lab is on how microbial growth quantitatively depends on resource availability. Our first progress on this question used laboratory growth measurements and a mathematical model of evolution to show that the response of microbial growth to nutrients can evolve independently of that nutrient’s concentration in the environment, meaning that microbes can evolve to grow rapidly in low concentrations even if they mainly live at high concentrations (Fink et al. 2023). Surprisingly, we also found that there is no evidence of tradeoffs between growth at low and high concentrations, as has often been claimed.
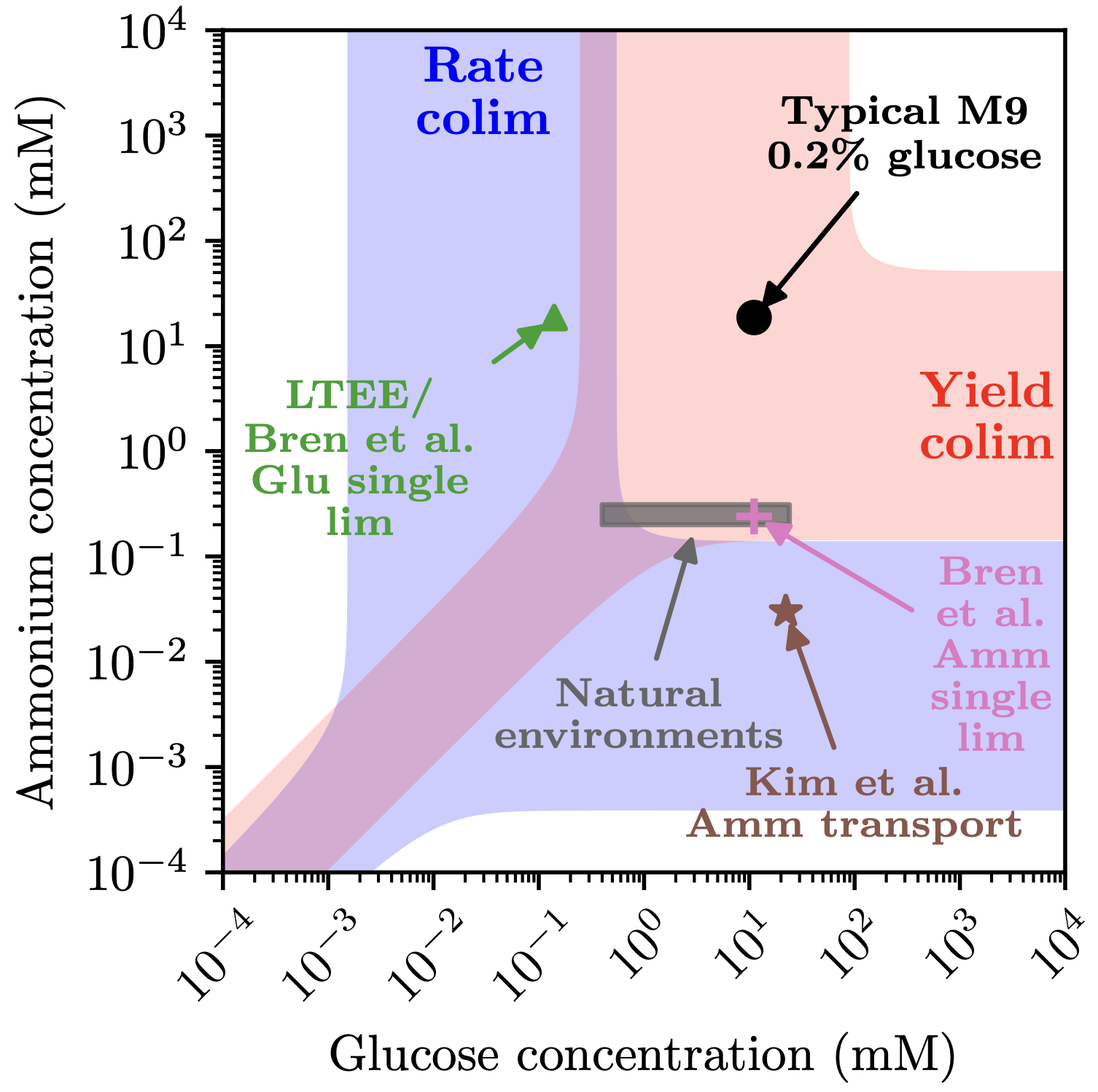
In collaboration with the lab of Noelle Held, a marine microbiologist and oceanographer at USC, we are now studying how the availabilities of multiple resources (e.g., a carbon source and a nitrogen source) jointly shape growth, known as colimitation (Held and Manhart 2024). We recently introduced a theoretical framework to quantify colimitation, which we used to show that microbial growth is readily colimited by multiple nutrients in both laboratory and natural environments (Held et al. 2024). Using a combination of laboratory and field experiments and mathematical modeling, we are currently working to understand the evolution of colimitation, cell physiology under colimitation (gene activity and proteome allocation), and how colimitation affects antibiotic sensitivity.
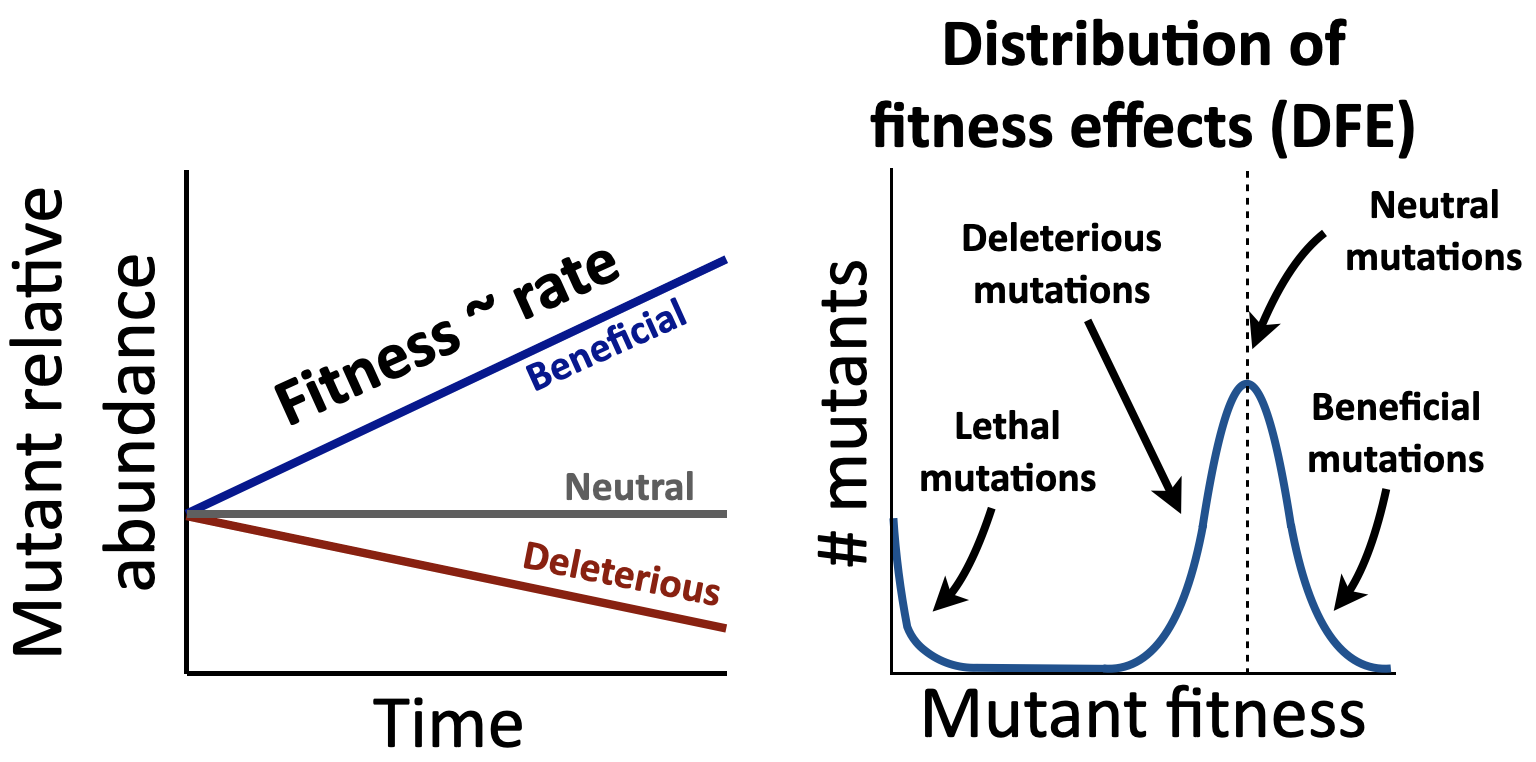
The supply of new mutations to microbial genomes is enormous, up to a trillion new mutations per day in a person's gut microbiome. Despite the growth of microbial genomic data, we still have little ability to predict the effects all these mutations have on fitness. This is a major challenge for microbiology since the distribution of fitness effects (DFE) — the set of fitness values for all spontaneous mutations available to a population — is a key input to predicting how a population will evolve. Our ability to predict evolution of microbes in different ecological environments is valuable for treating infectious diseases — for example, anticipating if and how fast a pathogen will evolve resistance to a treatment — as well as for designing personalized medicine based on an individual’s evolving microbiome.
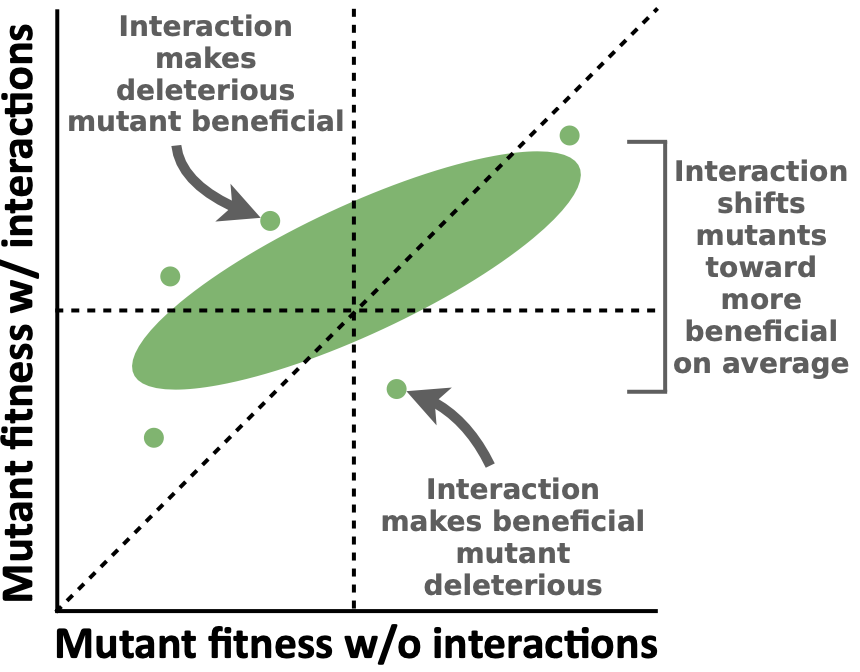
One factor that significantly complicates our ability to predict or measure mutant fitness is that it depends on the specific environmental context. A major feature of the environments microbes face is the presence of other microbial cells that a focal species interacts with, such as by competing for or exchanging nutrients (cross-feeding) (Corral López et al. 2026). Several recent studies have found that ecological interactions between microbes can significantly change mutant fitness and consequent adaptation (Gorter et al. 2020), meaning that our ability to predict microbial evolution from DFEs relies on understanding how mutant fitness depends on the ecological environment. Since we cannot empirically measure the fitness of mutations across all possible environments, it is crucial that we develop general principles for how mutant fitness changes across these environments to predict evolution.
Our current work in this research direction includes both experimental and computational components. First, we are working to measure the DFE under different types of cross-feeding interactions using a synthetic community of auxotroph strains that have been engineered to cross-feed specific nutrients, such as amino acids. We use mutant libraries consisting of random-barcoded (Jasinska et al. 2020) transposon-insertions, which mimic loss-of-function mutations that occur spontaneously in microbial genomes. Our goal is to empirically map the effect of different cross-feeding interactions on the DFE, so that we can predict whether these interactions would differentially alter evolutionary dynamics.
Second, we are using both genome-scale and coarse-grained mathematical models of these communities to generate hypotheses for the underlying mechanisms. In particular, we are exploring how the DFE varies under competitive vs. cross-feeding interactions, how changes in nutrient limitation brought on by cross-feeding alter mutant fitness, and what role metabolic cheating plays in determining mutant fitness under cross-feeding.
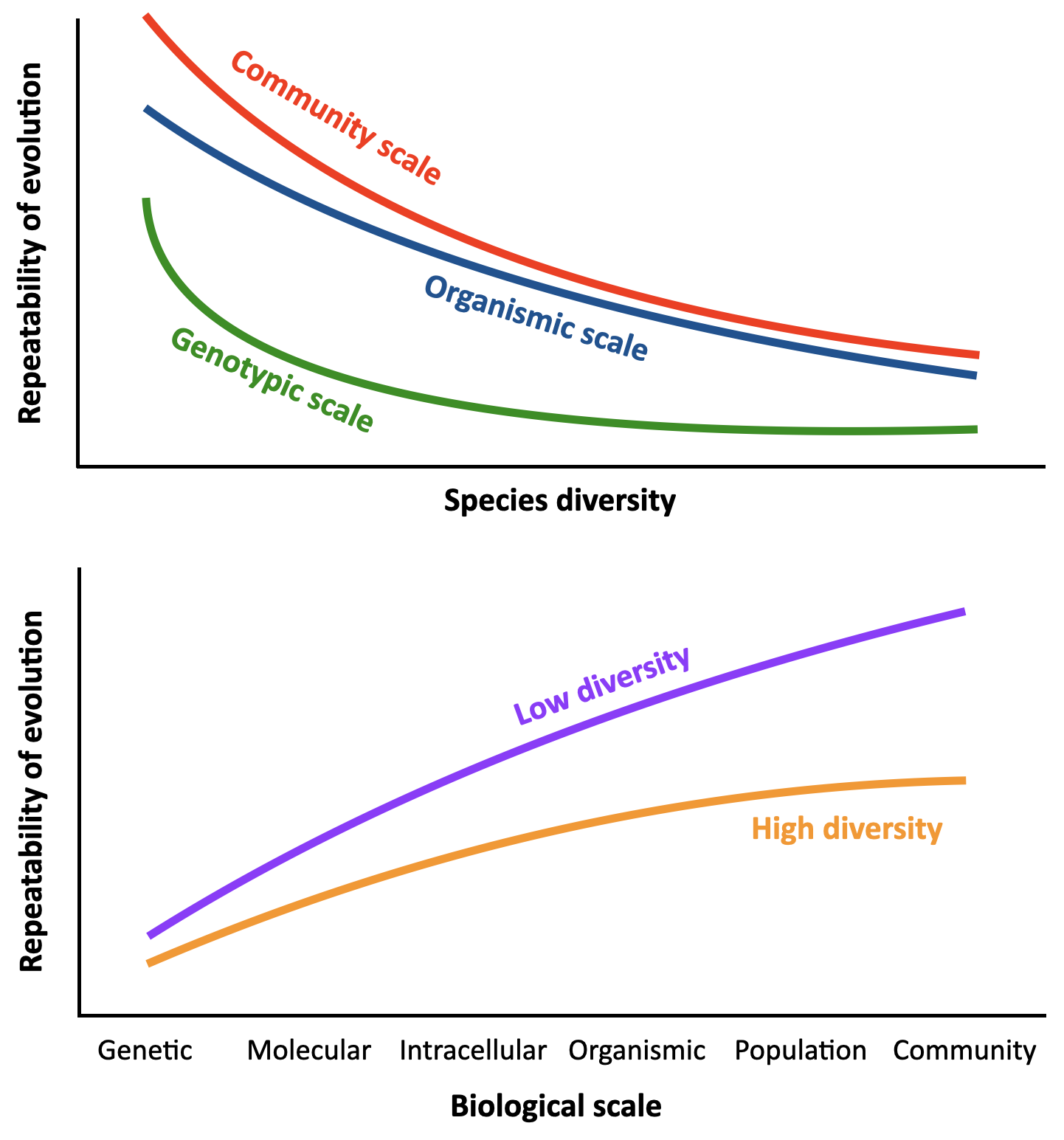
Predicting the future course of evolution is a critical problem for the study of infectious diseases, cancer, and climate change. While empirical data has shown that the predictability of evolution varies across biological scales — evolution usually being more predictable at large scales like traits of whole organisms, and less predictable at small scales like nucleotides in genomic DNA — it is unknown what biological scales are optimal for predicting evolutionary change. In particular, the role of ecological-scale properties is a major unknown for understanding evolutionary predictability in natural microbial ecosystems, which almost always consist of multiple species existing together.
Many human infections involve multispecies microbial communities, including Candida yeasts, a genus of fungal pathogens that is increasing evolving resistance to common antifungal drugs. The goal of this project — in collaboration with the labs of Daniel Charlebois (U. Alberta), an experimental biophysicist, and Meike Wortel (U. Amsterdam), an evolutionary systems biologist — is therefore to study how predictable is the evolution of multispecies communities of Candida yeasts, in the presence of the antifungal drug fluconazole. Using laboratory evolutionary experiments, multiscale models of cell physiology, and evolutionary theory, we aim to determine whether community diversity corresponds to less predictable evolutionary outcomes.

